Product information “Craniopharyngioma”
Clinical History
A 62-year-old woman presented with disorientation to time, place and person. Physical examination revealed no localised neurological signs. Radiological investigations revealed a space occupying lesion in the floor of the 3rd ventricle. Tissue was removed at surgery but the lesion could not be completely excised. Histology confirmed the diagnosis of Craniopharyngioma. Post-operatively the patient developed complex metabolic disturbances, probably hypothalamic in origin. She gradually deteriorated, and 10 weeks after admission she died following an episode of gastric aspiration.
Pathology
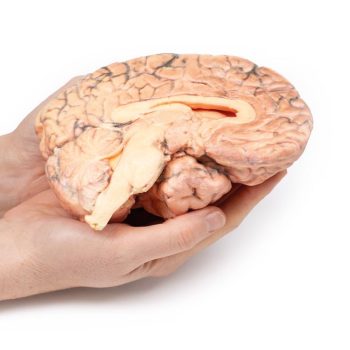
The brain has been sectioned in the sagittal plane, displaying the medial surface. A pink-grey, ovoid tumour measuring 2.5 x 1.5 cm on the cut surface is centred in the region of the hypothalamus. It is encapsulated except at its ventral pole where tissue has been removed at previous surgery, and the cut surface reveals a microcystic or spongy appearance. The tumour distorts the 3rd ventricle and extends to obliterate the Foramen of Munro. The optic chiasm is displaced caudally (arrow). Previous ventriculo-atrial shunting has prevented dilatation of the lateral ventricles despite this obstruction.
Further Information
Craniopharyngiomas constitute 1-3% of all brain tumours, and 5-10% in children, with a bimodal distribution favouring ages 5-14 years, and a second peak between ages 50-75 years. There is a higher incidence in Japan and parts of Africa. Craniopharyngiomas are epithelial tumours generally arising from the pituitary stalk. Other sites of origin include the sella turcica, optic system and third ventricle. There are frequently solid and cystic components, the latter containing cholesterol crystals. Craniopharyngiomas can be divided into two categories – adamantinomatous and papillary types, each with distinct histology and genetic alterations, although the prognostic significance of these types remains unclear.
Treatment includes surgical resection and radiation therapy (RT) to treat and post-surgical residual disease. Prognosis depends largely on tumour resection, control and treatment-related complications arising from local and endocrine and local sequelae.